User:Jhurley/sandbox
PFAS Destruction by Ultraviolet/Sulfite Treatment
The ultraviolet (UV)/sulfite based reductive defluorination process has emerged as an effective and practical option for generating hydrated electrons (eaq- ) which can destroy PFAS in water. It offers significant advantages for PFAS destruction, including significant defluorination, high treatment efficiency for long-, short-, and ultra-short chain PFAS without mass transfer limitations, selective reactivity by hydrated electrons, low energy consumption, low capital and operation costs, and no production of harmful byproducts. A UV/sulfite treatment system designed and developed by Haley and Aldrich (EradiFluorTM[1]) has been demonstrated in two field demonstrations in which it achieved near-complete defluorination and greater than 99% destruction of 40 PFAS analytes measured by EPA method 1633.
Related Article(s):
- Perfluoroalkyl and Polyfluoroalkyl Substances (PFAS)
- PFAS Ex Situ Water Treatment
- PFAS Sources
- PFAS Treatment by Electrical Discharge Plasma
- Supercritical Water Oxidation (SCWO)
- Photoactivated Reductive Defluorination - PFAS Destruction
Contributors: John Xiong, Yida Fang, Raul Tenorio, Isobel Li, and Jinyong Liu
Key Resources:
- Defluorination of Per- and Polyfluoroalkyl Substances (PFAS) with Hydrated Electrons: Structural Dependence and Implications to PFAS Remediation and Management[2]
- Accelerated Degradation of Perfluorosulfonates and Perfluorocarboxylates by UV/Sulfite + Iodide: Reaction Mechanisms and System Efficiencies[3]
- Destruction of Per- and Polyfluoroalkyl Substances (PFAS) in Aqueous Film-Forming Foam (AFFF) with UV-Sulfite Photoreductive Treatment[4]
- EradiFluorTM[1]
Introduction
The hydrated electron (eaq- ) can be described as an electron in solution surrounded by a small number of water molecules[5]. Hydrated electrons can be produced by photoirradiation of solutes, including sulfite, iodide, dithionite, and ferrocyanide, and have been reported in literature to effectively decompose per- and polyfluoroalkyl substances (PFAS) in water. The hydrated electron is one of the most reactive reducing species, with a standard reduction potential of about −2.9 volts. Though short-lived, hydrated electrons react rapidly with many species having more positive reduction potentials[5].
Among the electron source chemicals, sulfite (SO32−) has emerged as one of the most effective and practical options for generating hydrated electrons to destroy PFAS in water. The mechanism of hydrated electron production in a sulfite solution under ultraviolet is shown in Equation 1 (UV is denoted as hv, SO3•- is the sulfur trioxide radical anion):
- Equation 1:
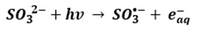
- Equation 1:
The hydrated electron has demonstrated excellent performance in destroying PFAS such as perfluorooctanoic acid (PFOA), perfluorooctanesulfonic acid (PFOS)[6] and GenX[7]. Mechanisms include cleaving carbon-to-fluorine (C-F) bonds (i.e., hydrogen/fluorine atom exchange) and chain shortening (i.e., decarboxylation, hydroxylation, elimination, and hydrolysis)[2].
PFAS Background
PFAS are a broad class of chemicals with highly variable chemical structures[8][9][10]. One characteristic feature of PFAS is that they are fluorosurfactants, distinct from more traditional hydrocarbon surfactants[9][11][12][13]. Fluorosurfactants typically have a fully or partially fluorinated, hydrophobic tail with ionic (cationic, zwitterionic, or anionic) head group that is hydrophilic[9][10]. The hydrophobic tail and ionic head group mean PFAS are very stable at hydrophobic adsorption interfaces when present in the aqueous phase[14]. Examples of these interfaces include naturally occurring organic matter in soils and the air-water interface in the vadose zone[15][16][17][18][19]. Their strong adsorption to both soil organic matter and the air-water interface is a major contributor to elevated concentrations of PFAS observed in the upper 5 feet of the soil column[20][21]. While several other PFAS partitioning processes exist[11], adsorption to solid phase soils and air-water interfaces are the two primary processes present at nearly all PFAS sites[22]. The total PFAS mass obtained from a vadose zone soil sample contains the solid phase, air-water interfacial, and aqueous phase PFAS mass, which can be converted to porewater concentrations using Equation 1[23].
- Equation 1:
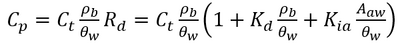
- Equation 1:
Where Cp is the porewater concentration, Ct is the total PFAS concentration, ρb is the bulk density of the soil, θw is the volumetric water content, Rd is the PFAS retardation factor, Kd is the solid phase adsorption coefficient, Kia is the air-water interfacial adsorption coefficient, and Aaw is the air-water interfacial area. The air-water interfacial area of the soil is primarily a function of both the soil properties and the degree of volumetric water saturation in the soil. There are several methods of estimating air-water interfacial areas including thermodynamic functions based on the soil moisture retention curve. However, the thermodynamic function has been shown to underestimate air-water interfacial area[24], and must typically be scaled using empirical scaling factors. An empirical method recently developed to estimate air-water interfacial area is presented in Equation 2[24].
- Equation 2:
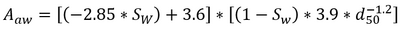
- Equation 2:
Where Sw is the water phase saturation as a ratio of the water content over the volumetric soil porosity, and d50 is the median grain diameter.
Lysimeters Background

Lysimeters, generally speaking, refer to instruments which collect water from unsaturated soils[27][28]. However, there are multiple types of lysimeters which can be employed in field or laboratory settings. There are three primary types of lysimeters relevant to PFAS listed here and shown in Figure 1a-d.
- Suction Lysimeters (Figure 1a,b): These lysimeters are the most relevant for PFAS sampling and are the majority of discussion in this article. These lysimeters operate by extracting liquid from the unsaturated vadose zone by applying negative suction pressure at the sampling head[29][25][30]. The sampling head is typically constructed of porous ceramic or stainless steel. A PVC case or stainless-steel case is attached to the sampling head and extends upward above the ground surface. Suction lysimeters are typically installed between 1 and 9 feet below ground surface, but can extend as deep as 40-60 feet in some cases[29]. Shallow lysimeters (< 10 feet) are typically installed using a hand auger. For ceramic lysimeters, a silica flour slurry should be placed at the base of the bore hole and allowed to cover the ceramic head before backfilling the hole partially with natural soil. Once the hole is partially backfilled with soil to cover the sampling head, the remainder of the casing should be sealed with hydrated bentonite chips. When sampling events occur, suction is applied at the ground surface using a rubber gasket seal and a hand pump or electric pump. After sufficient porewater is collected (the time for which can vary greatly based on the soil permeability and moisture content), the seal can be removed and a peristaltic pump used to extract liquid from the lysimeter.
- Field Lysimeters (Figure 1c): These large lysimeters can be constructed from plastic or metal sidings. They can range from approximately 2 feet in diameter to as large as several meters in diameter[27]. Instrumentation such as soil moisture probes and tensiometers, or even multiple suction lysimeters, are typically placed throughout the lysimeter to measure the movement of water and determine characteristic soil moisture release curves[31][32][26][33][34]. Water is typically collected at the base of the field lysimeter to determine net recharge through the system. These field lysimeters are intended to represent more realistic, intermediate scale conditions of field systems.
- Drainage Lysimeters (Figure 1d): Also known as a “wick” lysimeter, these lysimeters typically consist of a hollow cup attached to a spout which protrudes above ground to relieve air pressure from the system and act as a sampling port. The hollow cup typically has filters and wicking devices at the base to collect water from the soil. The cup is filled with natural soil and collects water as it percolates through the vadose zone. These lysimeters are used to directly monitor net recharge from the vadose zone to the groundwater table and could be useful in determining PFAS mass flux.
Analysis of PFAS Concentrations in Soil and Porewater
| Site | PFAS | Field Porewater Concentration (μg/L) |
Lab Core Porewater Concentration (μg/L) |
Predicted Porewater Concentration (μg/L) |
|---|---|---|---|---|
| Site A | PFOS | 6.2 ± 3.4 | 3.0 ± 0.37 | 6.6 ± 3.3 |
| Site B | PFOS | 2.2 ± 2.0 | 0.78 ± 0.38 | 2.8 |
| Site C | PFOS | 13 ± 4.1 | 680 ± 460 | 164 ± 75 |
| 8:2 FTS | 1.2 ± 0.46 | 52 ± 13 | 16 ± 6.0 | |
| PFHpS | 0.36 ± 0.051 | 2.9 ± 2.0 | 5.9 ± 3.4 |


Schaefer et al.[25] measured PFAS porewater concentrations with field and laboratory suction lysimeters across several sites. Intact cores from the site were collected for soil water extraction using laboratory lysimeters. The lysimeters were used to directly compare field derived measurements of PFAS concentration in the mobile porewater phase. Results from measurements are for four sites presented in Figure 2.
Data from sites A and B showed reasonably good agreement (within ½ order of magnitude) for most PFAS measured in the systems. At site C, more hydrophobic constituents (> C6 PFAS) tended to have higher concentrations in the lab core than the field site while less hydrophobic constituents (< C6) had higher concentrations in the field than lab cores. Site D showed substantially greater (1 order of magnitude or more) PFAS concentrations measured in the laboratory-collected porewater sample compared to what was measured in the field lysimeters. This discrepancy for the Site D soil can likely be attributed to soil heterogeneity (as indicated by ground penetrating radar) and the fact that the soil consisted of back-filled materials rather than undisturbed native soils.
Site C showed elevated PFAS concentrations in the laboratory collected porewater for the more surface-active compounds. This increase was attributed to the soil wetting that occurred at the bench scale, which was reasonably described by the model shown in Equations 1 and 2 (see Table 1[35]). Equations 1 and 2 were also used to predict PFAS porewater concentrations (using porous cup lysimeters) in a highly instrumented test cell[26](Figure 3). The ability to predict soil concentrations from recurring porewater samples is critical to the practical application of lysimeters in field settings[35].
Results from suction lysimeters studies and field lysimeter studies show that PFAS concentrations in porewater predicted from soil concentrations using Equations 1 and 2 generally have reasonable agreement with measured in situ porewater data when air-water interfacial partitioning is considered. Results show that for less hydrophobic components like PFOA, the impact of air-water interfacial adsorption is less significant than for highly hydrophobic components like PFOS. The soil for the field lysimeter in Figure 3 was a sandy soil with a relatively low air-water interfacial area. The effect of air-water interfacial partitioning is expected to be much more significant for a greater range of PFAS in soils with high capillary pressure (i.e. silts/clays) with higher associated air-water interfacial areas[24][36][37].
Summary and Recommendations
The majority of research with lysimeters for PFAS site investigations has been done using porous cup suction lysimeters[29][35][25][30]. Porous cup suction lysimeters are advantageous because they can be routinely sampled or sampled after specific wetting or drying events much like groundwater wells. This sampling is easier and more efficient than routinely collecting soil samples from the same locations. Co-locating lysimeters with soil samples is important for establishing the baseline soil concentration levels at the lysimeter location and developing correlations between the soil concentrations and the mobile porewater concentration[29]. Appropriate standard operation procedures for lysimeter installation and operation have been established and have been reviewed in recent literature[29][25]. Lysimeters should typically be installed near the source area and just above the maximum groundwater level elevation to obtain accurate results of porewater concentrations year round. Depending upon the geology and vertical PFAS distribution in the soil, multilevel lysimeter installations should also be considered.
Results from several lysimeters studies across multiple field sites and modelling analysis has shown that lysimeters can produce reasonable results between field and laboratory studies[25][26][33]. Transient effects of wetting and drying as well as media heterogeneity affects appear to be responsible for some variability and uncertainty in lysimeter based PFAS measurements in the vadose zone. These mobile porewater concentrations can be coupled with effective recharge estimates and simplified modelling approaches to determine mass flux from the vadose zone to the underlying groundwater[38][39][23][40][41].
Future research opportunities should address the current key uncertainties related to the use of lysimeters for PFAS investigations, including:
- Collect larger datasets of PFAS concentrations to determine how transient wetting or drying periods and media type affect PFAS concentrations in the mobile porewater. Some research has shown that non-equilibrium processes can occur in the vadose zone, which can affect grab sample concentration in the porewater at specific time periods.
- More work should be done with flux averaging lysimeters like the drainage cup or wicking lysimeter. These lysimeters can directly measure net recharge and provide time averaged concentrations of PFAS in water over the sampling period. However, there is little work detailing their potential applications in PFAS research, or operational considerations for their use in remedial investigations for PFAS.
- Lysimeters should be coupled with monitoring of wetting and drying in the vadose zone using in situ soil moisture sensors or tensiometers and groundwater levels. Direct measurements of soil saturation at field sites are vital to directly correlate porewater concentrations with soil concentrations. Similarly, groundwater level fluctuations can inform net recharge estimates. By collecting these data we can continue to improve partitioning and leaching models which can relate porewater concentrations to total PFAS mass in soils and PFAS leaching at field sites.
- Comparisons of various bench-scale leaching or desorption tests to field-based lysimeter data are recommended. The ability to correlate field measurements of PFAS concentrations with estimates of leaching from laboratory studies would provide a powerful method to empirically estimate PFAS leaching from field sites.
References
- ^ 1.0 1.1 Haley and Aldrich, Inc. (commercial business), 2024. EradiFluor. Comercial Website
- ^ 2.0 2.1 Bentel, M.J., Yu, Y., Xu, L., Li, Z., Wong, B.M., Men, Y., Liu, J., 2019. Defluorination of Per- and Polyfluoroalkyl Substances (PFASs) with Hydrated Electrons: Structural Dependence and Implications to PFAS Remediation and Management. Environmental Science and Technology, 53(7), pp. 3718-28. doi: 10.1021/acs.est.8b06648 Open Access Article
- ^ Liu, Z., Chen, Z., Gao, J., Yu, Y., Men, Y., Gu, C., Liu, J., 2022. Accelerated Degradation of Perfluorosulfonates and Perfluorocarboxylates by UV/Sulfite + Iodide: Reaction Mechanisms and System Efficiencies. Environmental Science and Technology, 56(6), pp. 3699-3709. doi: 10.1021/acs.est.1c07608 Open Access Article
- ^ Tenorio, R., Liu, J., Xiao, X., Maizel, A., Higgins, C.P., Schaefer, C.E., Strathmann, T.J., 2020. Destruction of Per- and Polyfluoroalkyl Substances (PFASs) in Aqueous Film-Forming Foam (AFFF) with UV-Sulfite Photoreductive Treatment. Environmental Science and Technology, 54(11), pp. 6957-67. doi: 10.1021/acs.est.0c00961
- ^ 5.0 5.1 Buxton, G.V., Greenstock, C.L., Phillips Helman, W., Ross, A.B., 1988. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (⋅OH/⋅O-) in Aqueous Solution. Journal of Physical and Chemical Reference Data, 17(2), pp. 513-886. doi: 10.1063/1.555805
- ^ Gu, Y., Liu, T., Wang, H., Han, H., Dong, W., 2017. Hydrated Electron Based Decomposition of Perfluorooctane Sulfonate (PFOS) in the VUV/Sulfite System. Science of The Total Environment, 607-608, pp. 541-48. doi: 10.1016/j.scitotenv.2017.06.197
- ^ Bao, Y., Deng, S., Jiang, X., Qu, Y., He, Y., Liu, L., Chai, Q., Mumtaz, M., Huang, J., Cagnetta, G., Yu, G., 2018. Degradation of PFOA Substitute: GenX (HFPO–DA Ammonium Salt): Oxidation with UV/Persulfate or Reduction with UV/Sulfite? Environmental Science and Technology, 52(20), pp. 11728-34. doi: 10.1021/acs.est.8b02172
- ^ Moody, C.A., Field, J.A., 1999. Determination of Perfluorocarboxylates in Groundwater Impacted by Fire-Fighting Activity. Environmental Science and Technology, 33(16), pp. 2800-2806. doi: 10.1021/es981355+
- ^ 9.0 9.1 9.2 Moody, C.A., Field, J.A., 2000. Perfluorinated Surfactants and the Environmental Implications of Their Use in Fire-Fighting Foams. Environmental Science and Technology, 34(18), pp. 3864-3870. doi: 10.1021/es991359u
- ^ 10.0 10.1 Glüge, J., Scheringer, M., Cousins, I.T., DeWitt, J.C., Goldenman, G., Herzke, D., Lohmann, R., Ng, C.A., Trier, X., Wang, Z., 2020. An Overview of the Uses of Per- and Polyfluoroalkyl Substances (PFAS). Environmental Science: Processes and Impacts, 22(12), pp. 2345-2373. doi: 10.1039/D0EM00291G Open Access Article
- ^ 11.0 11.1 Brusseau, M.L., 2018. Assessing the Potential Contributions of Additional Retention Processes to PFAS Retardation in the Subsurface. Science of The Total Environment, 613-614, pp. 176-185. doi: 10.1016/j.scitotenv.2017.09.065 Open Access Manuscript
- ^ Dave, N., Joshi, T., 2017. A Concise Review on Surfactants and Its Significance. International Journal of Applied Chemistry, 13(3), pp. 663-672. doi: 10.37622/IJAC/13.3.2017.663-672 Open Access Article
- ^ García, R.A., Chiaia-Hernández, A.C., Lara-Martin, P.A., Loos, M., Hollender, J., Oetjen, K., Higgins, C.P., Field, J.A., 2019. Suspect Screening of Hydrocarbon Surfactants in Afffs and Afff-Contaminated Groundwater by High-Resolution Mass Spectrometry. Environmental Science and Technology, 53(14), pp. 8068-8077. doi: 10.1021/acs.est.9b01895
- ^ Krafft, M.P., Riess, J.G., 2015. Per- and Polyfluorinated Substances (PFASs): Environmental Challenges. Current Opinion in Colloid and Interface Science, 20(3), pp. 192-212. doi: 10.1016/j.cocis.2015.07.004
- ^ Schaefer, C.E., Culina, V., Nguyen, D., Field, J., 2019. Uptake of Poly- and Perfluoroalkyl Substances at the Air–Water Interface. Environmental Science and Technology, 53(21), pp. 12442-12448. doi: 10.1021/acs.est.9b04008
- ^ Lyu, Y., Brusseau, M.L., Chen, W., Yan, N., Fu, X., Lin, X., 2018. Adsorption of PFOA at the Air–Water Interface during Transport in Unsaturated Porous Media. Environmental Science and Technology, 52(14), pp. 7745-7753. doi: 10.1021/acs.est.8b02348
- ^ Costanza, J., Arshadi, M., Abriola, L.M., Pennell, K.D., 2019. Accumulation of PFOA and PFOS at the Air-Water Interface. Environmental Science and Technology Letters, 6(8), pp. 487-491. doi: 10.1021/acs.estlett.9b00355
- ^ Li, F., Fang, X., Zhou, Z., Liao, X., Zou, J., Yuan, B., Sun, W., 2019. Adsorption of Perfluorinated Acids onto Soils: Kinetics, Isotherms, and Influences of Soil Properties. Science of The Total Environment, 649, pp. 504-514. doi: 10.1016/j.scitotenv.2018.08.209
- ^ Nguyen, T.M.H., Bräunig, J., Thompson, K., Thompson, J., Kabiri, S., Navarro, D.A., Kookana, R.S., Grimison, C., Barnes, C.M., Higgins, C.P., McLaughlin, M.J., Mueller, J.F., 2020. Influences of Chemical Properties, Soil Properties, and Solution pH on Soil–Water Partitioning Coefficients of Per- and Polyfluoroalkyl Substances (PFASs). Environmental Science and Technology, 54(24), pp. 15883-15892. doi: 10.1021/acs.est.0c05705 Open Access Article
- ^ Cite error: Invalid
<ref>tag; no text was provided for refs namedBrusseauEtAl2020 - ^ Cite error: Invalid
<ref>tag; no text was provided for refs namedBiglerEtAl2024 - ^ Brusseau, M.L., Yan, N., Van Glubt, S., Wang, Y., Chen, W., Lyu, Y., Dungan, B., Carroll, K.C., Holguin, F.O., 2019. Comprehensive Retention Model for PFAS Transport in Subsurface Systems. Water Research, 148, pp. 41-50. doi: 10.1016/j.watres.2018.10.035
- ^ 23.0 23.1 Cite error: Invalid
<ref>tag; no text was provided for refs namedBrusseauGuo2022 - ^ 24.0 24.1 24.2 Brusseau, M.L., 2023. Determining Air-Water Interfacial Areas for the Retention and Transport of PFAS and Other Interfacially Active Solutes in Unsaturated Porous Media. Science of The Total Environment, 884, Article 163730. doi: 10.1016/j.scitotenv.2023.163730 Open Access Article
- ^ 25.0 25.1 25.2 25.3 25.4 25.5 Cite error: Invalid
<ref>tag; no text was provided for refs namedSchaeferEtAl2024 - ^ 26.0 26.1 26.2 26.3 26.4 Cite error: Invalid
<ref>tag; no text was provided for refs namedSchaeferEtAl2023 - ^ 27.0 27.1 Cite error: Invalid
<ref>tag; no text was provided for refs namedMeissnerEtAl2020 - ^ Cite error: Invalid
<ref>tag; no text was provided for refs namedRogersMcConnell1993 - ^ 29.0 29.1 29.2 29.3 29.4 Cite error: Invalid
<ref>tag; no text was provided for refs namedCostanzaEtAl2025 - ^ 30.0 30.1 Cite error: Invalid
<ref>tag; no text was provided for refs namedQuinnanEtAl2021 - ^ Cite error: Invalid
<ref>tag; no text was provided for refs namedStannard1992 - ^ Cite error: Invalid
<ref>tag; no text was provided for refs namedWintonWeber1996 - ^ 33.0 33.1 Cite error: Invalid
<ref>tag; no text was provided for refs namedSchaeferEtAl2022 - ^ van Genuchten, M.Th. , 1980. A Closed‐form Equation for Predicting the Hydraulic Conductivity of Unsaturated Soils. Soil Science Society of America Journal, 44(5), pp. 892-898. doi: 10.2136/sssaj1980.03615995004400050002x
- ^ 35.0 35.1 35.2 35.3 Cite error: Invalid
<ref>tag; no text was provided for refs namedAndersonEtAl2022 - ^ Peng, S., Brusseau, M.L., 2012. Air-Water Interfacial Area and Capillary Pressure: Porous-Medium Texture Effects and an Empirical Function. Journal of Hydrologic Engineering, 17(7), pp. 829-832. doi: 10.1061/(asce)he.1943-5584.0000515
- ^ Brusseau, M.L., Peng, S., Schnaar, G., Costanza-Robinson, M.S., 2006. Relationships among Air-Water Interfacial Area, Capillary Pressure, and Water Saturation for a Sandy Porous Medium. Water Resources Research, 42(3), Article W03501, 5 pages. doi: 10.1029/2005WR004058 Free Access Article
- ^ Cite error: Invalid
<ref>tag; no text was provided for refs namedAnderson2021 - ^ Cite error: Invalid
<ref>tag; no text was provided for refs namedStultsEtAl2024 - ^ Stults, J.F., Schaefer, C.E., MacBeth, T., Fang, Y., Devon, J., Real, I., Liu, F., Kosson, D., Guelfo, J.L., 2025. Laboratory Validation of a Simplified Model for Estimating Equilibrium PFAS Mass Leaching from Unsaturated Soils. Science of The Total Environment, 970, Article 179036. doi: 10.1016/j.scitotenv.2025.179036
- ^ Smith, J. Brusseau, M.L., Guo, B., 2024. An Integrated Analytical Modeling Framework for Determining Site-Specific Soil Screening Levels for PFAS. Water Research, 252, Article121236. doi: 10.1016/j.watres.2024.121236